

Если вкратце, на сегодняшний день это лучший показатель биовозраста, который точнее паспорта отражает, сколько нам осталось жить. Что такое эпигенетические часы я когда-то уже писал. Потому что я считаю, что старение — это эпигенетическая программа. Почему эпичасы так важны? Да без проблем, готов считать этот тезис своим «Символом веры». Кто-то называет это моё убеждение религиозным. Ну или «Символом Юры» — верую, что старение есмь эпигенетическая программа и чаю её взлом.
В котором особое место отведено тем самым эпигенетическим часам метилирования. Ведь верую я в это не просто так, а на основании всего уместившегося на сегодняшний день в моей голове массива данных. Особое оно из-за того, что с возрастом у млекопитающих метилирование ДНК не просто стохастически «вымывается», как можно было бы предположить при случайной природе этого процесса, а часто наоборот усиливается, что обычно коррелирует со снижением экспрессии различных нужных генов (нужных, потому что если бы они не были нужными, они были бы выключены изначально, хотя бы после полового созревания, а не плавно выключались по мере старения).
Чуть дальше я вернусь к этой теме и приведу несколько работ, показавших такое снижение у разных видов. Вообще наблюдения возрастного снижения экспрессии нужных генов всплывают в сфере изучения биологических механизмов старения постоянно.
Это приводит к гиперактивации врождённого иммунитета, то есть к пресловутому «инфламэйджингу» — стерильному возрастному воспалению, — которое поголовно наблюдается у пожилых людей и животных.
Почему наш организм разрешает транспозонам выйти на волю после определённого возраста? При этом наблюдается и обратный процесс: некоторые совсем ненужные нам гены, такие как вредоносные ретротраспозоны, в раннем возрасте не просто выключены, а находятся за семью печатями, но к определённому моменту печати почему-то распечатываются и эта «пятая колонна» начинает свою активность, подвергая бомбардировке наши гены своими копиями. А для половых клеток, которые вынуждены обнулять своё метилирование после оплодотворения, у организма есть ещё один механизм защиты от вредоносных «генов-рукиножниц» — piRNA. Ведь на примере других генов мы видим, что он вполне способен поддерживать или даже повышать метилирование ДНК с возрастом. Что это, равнодушное попустительство генов по отношению к выполнившей свою репродукционную роль «расходной соме» или целенаправленный феноптоз? Так почему же на закате репродуктивного возраста транспозонам дозволяется выйти из-под этого контроля?
Объясню почему. Мне видится, что второе. Напомню, что у некоторых видов термитов репродуктивная каста живёт в 80-120 раз дольше своих идентичных близнецов, пошедших по рабочей жизненной стезе. В природе есть очень показательный пример социальных животных: пчёл, муравьёв, термитов. Так вот, в одной работе авторы показали, что у маток та самая мегазащита от транспозонов, piRNA, активна на протяжении многих лет, а у рабочих особей она снижается за считанные месяцы:
«Поразительно, что гены из пути PIWI-взаимодействующей РНК (piRNA), которые, как известно, заставляют молчать [транспозоны] в зародышевой линии многоклеточных животных, были подавлены только у старых рабочих, но не у королевской касты».
Ну да ладно, транспозоны — тема для отдельного поста, а в этом я хочу вернуться к часам метилирования и подробно разобрать одну интересную работу от группы Вадима Гладышева, в которой они досконально проанализировали, что происходит с метилированием по мере взросления и старения (мышей). Для этого они снимали мышиный профиль метилирования аж в 16-и временных точках — то есть, практически, через каждые 2 месяца короткой мышиной жизни. И не просто «профиль метилирования» какой-нибудь сотни-другой сайтов, а изучили целых 800 000 различных сайтов метилирования, из которых чуть более 20% значимо менялись с возрастом.
В той исходной работе, во-первых, были созданы мышиные часы метилирования, а во-вторых, было показано, что эти часы замедляются от различных уже известных интервенций по продлению мышиной жизни (ограничение калорий и всё такое). Кстати, стоит отметить, что для своей статьи авторы использовали данные по возрастным изменениям мышиного метилома, полученными своими коллегами из той же лаборатории в более ранней работе. Кстати, те мышиные часы метилирования Гладышева в своё время похвалил сам Стив Хорват, самый известный эпигенетический часовщик.
Важная статья от группы Гладышева
За что же, в первую очередь, зацепился мой «верующий» глаз в этой работе? Вот за эти строки:
В возрастающей группе наиболее часто встречающиеся каскады были связаны с процессами развития. Среди промоторов, мы обнаружили 102 генных каскада, значимо ассоциированных с возрастным снижением метилирования, и 1162 каскада, ассоциированных с его увеличением. Среди каскадов со снижением метилирования были каскады, связанные с репарацией ДНК, иммунными процессами и воспалением. Были также значительно обогащены каскады, связанные со старением и продлевающими жизнь интервенциями, такие как реакция на факторы роста, инсулиноподобный фактор роста и TGFβ, каскад MAPK, сигнальные пути WNT и Notch, регуляция стволовых клеток, ответ на эстрадиол, пути регуляции метаболизма жирных кислот, а также пути регуляции транскрипции.
В генах мы наблюдали схожие паттерны по сравнению с промоторами. Мы также исследовали метилирование самих генов и обнаружили 39 значимых каскада, которые по мере старения теряли метилирование, и 987 каскада, чьё метилирование возрастало. Каскады с повышенным метилированием включали различные каскады развития, а также каскады, связанные со старением, включая регуляция клеточного старения и сенесценции, ответ на такие факторы роста как TGFβ, пролиферацией и дифференцировкой стволовых клеток, каскад MAPK, передачу сигналов WNT, Notch, и каскады метаболизма жирных кислот. Каскады с уменьшающимся метилированием ДНК включали репарацию ДНК, иммунную функцию и связанные с воспалением каскады. Кроме того, [в этой группе] был обнаружен путь, включающий само метилирование ДНК, включая ген DNMT1.
То есть что мы видим? Что тех генных каскадов, где метилирование с возрастом возрастает (что, напомню, обычно коррелирует со снижением генной экспрессии) в десятки раз больше, чем тех, где снижается. При этом интересно видеть, что повышается метилирование и некоторых «шестерёнок» самого механизма метилирования, таких как DNMT1, одна из функций которой — дублировать метильные метки при репликации ДНК с исходной нити на строящуюся. Может быть, именно поэтому разные вредоносные элементы, которые в раннем возрасте находятся под плотным репрессивным контролем, постепенно «выходят на свободу», по мере того как падает эффективность систем поддержания их метилирования. При этом вдвойне странно видеть, что на новое заметилирование ранее активных генов у организма почему-то сил и внимания хватает. Вернее, странно для тех, кто не верит, что гены могут целенаправленно пытаться этот самый организм убить. Для нас же, Свидетелей Феноптоза, всё логично.
Что авторы этой работы — убеждённые стохастики, то есть люди, считающие старение не программой, а случайным накоплением повреждений. Но знаете, что забавно? Вот как интерпретирует увиденное один из соавторов статьи, Александр Тышковский:
Когда мы анализировали конкретные изменения профиля метилирования с возрастом, увидели 2 основных паттерна. По поводу часов [метилирования] и их детерминированности.
Сайты, абсолютно заметилированные, с возрастом начинают деметилироваться, и наоборот. Первый — рост энтропии (объясняет изменение статуса метилирования с возрастом для более половины сайтов). Да, безусловно. Детерминированы ли эти изменения? Нет, ведь объясняются чистой стохастикой: уровень метилирования полностью заметилированных сайтов не может стать еще выше, он может только падать (что и происходит), то же самое с противоположной стороны. Запрограммированы ли они? Другими словами, их статус метилирования движется в направлении среднего с разных сторон как следствие падения контроля за статусом метилирования.
Программа ли это? Второй паттерн — падение метилирования (и рост активации) генов, связанных с поддержанием организма и исправлением повреждений (например, гены репарации ДНК и иммунного ответа). Но не старения, а исправления повреждений, которые увеличиваются с возрастом. Да. Но это не значит, что есть программа поломки автомобиля. Когда автомобиль неисправен, хозяин “запрограммировано” отвозит его в автомастерскую.
Опять энтропия
Ох уж эта энтропия. То и дело всплывает в разговорах о старении. Ну да, да, по мере старения энтропия возрастает — причём почему-то с очень разной скоростью в разных видах — но, как я уже писал, не потому что какой-то физический закон её обязывает это делать, а потому что гены ей это разрешают, а то и всячески помогают. Когда генам нужно энтропию организма уменьшать, они с этим прекрасно справляются — во время эмбриогенеза, например, или по мере полового созревания. Некоторые виды даже отрезанные конечности во взрослом возрасте могут отращивать, включая голову, как планарии. Но об этом феномене будет отдельный пост, а мы давайте вернёмся к тезису Александра об энтропии. На основании каких данных он сделал такие выводы?
Судя по графикам в его статье, на основании вот этих данных для мышей:

(с) Shannon entropy of the sites that significantly change (or do not change) with age.
(d) Shannon entropy of the sites that significantly increase and decrease with age.
…и вот этих данных для людей:

Age-related changes in entropy of the human DNA methylome.
(A) Shannon entropy of the sites that significantly change (or do not change) with age in 651 human samples from the age of 19 to 101 years. Permutation test was performed to assess the difference in entropy between changing and non-changing groups.
(B) Shannon entropy of the sites that significantly increase and decrease with age in 651 human samples from the age of 19 to 101 years. Permutation test was performed to assess the difference in entropy for increasing and decreasing groups.
Наоборот, средний уровень энтропии сайтов метилирования на удивление стабилен с возрастом. Сказать честно, я не вижу тут особого роста энтропии, особенно у людей. Более того, у тех человеческих сайтов, где метилирование с возрастом снижается, энтропия вообще падает (самый нижний график, синие точки).
Сначала это увидели у близнецов:
Тут стоит упомянуть, что показанное на графиках выше возрастное снижение энтропии нашего метилирования вполне соотносится с весьма любопытным явлением: после 75–80 лет разброс в эпигенетике у разных людей вместо того чтобы монотонно нарастать, начинает наоборот снижаться.
В исследованиях близнецов вариация в глобальном метилировании генома постепенно возрастала до 75 лет, но демонстрировала тенденцию к снижению в самой старой группе близнецов (76–88 лет)».
https://www.ncbi.nlm.nih.gov/pubmed/22621408 «Есть экспериментальные данные, свидетельствующие о том, что эпигенетические колебания могут перестать расходиться у очень старых людей.
А потом и не только у них:
«Возрастная эпигенетическая дивергенция, как это ни парадоксально, переходит в конвергенцию на более поздних этапах жизни.
1186/s13059-016-0946-8 Мы находим, что ткани мозга пожилых людей (> 75 лет) становятся более похожими друг на друга, как эпигенетически, так и транскрипционно, по сравнению с более молодыми людьми».
https://genomebiology.biomedcentral.com/articles/10.
Позволю себе немного лирики — в принципе, что-то подобное мы наблюдаем и в жизни. Разброс в уровнях здоровья у людей в когорте 50-60 лет достаточно велик: в этом возрасте ещё есть много живчиков, но вот в 80 лет уже практически все люди довольно дряхлые. Может быть, именно это отражает динамика метилирования. На самом деле, это очень грустно, потому что показывает, что как бы мы не старались сохранить себе молодость ведя здоровый образ жизни, похоже, что к определённому возрасту мы всё равно догоним своих менее дисциплинированных ровесников по уровню дряхлости. То есть наши траектории старения сойдутся.
Кстати, забавно, что схождение траекторий старения также прослеживается между богатыми и бедными — деньги могут значительно увеличить ваши шансы дожить до 75, но не до 100 лет:

Ладно, вернёмся к нашим мышам и их энтропии. Последнее, что я хотел сказать на эту тему, это то, что, если приглядеться, видно, что на протяжении подавляющего большинства мышиной жизни уровень энтропии их метилирования остаётся примерно неизменным — практически до 2-х лет жизни (а средняя продолжительность жизни мыши — 2,5 года). Вот те же мышиные данные авторов, только на более наглядных графиках:

Age-related increases in entropy of the DNA methylome.
(A) Shannon entropy of 141 C57BL/6 mouse samples, calculated for every site.
(B) Shannon entropy of the same samples, calculated only for the sites that significantly change with age.
Если закрыть рукой правые части графиков (после 22–24 месяцев), то практически никакого роста и не видно.
Метилирование с возрастом
Ну да бог с ней, с энтропией. Давайте более пристально присмотримся к самому метилированию. Насколько верен тезис, что изменения метилирования “объясняются чистой стохастикой: уровень метилирования полностью заметилированных сайтов не может стать еще выше, он может только падать (что и происходит), то же самое с противоположной стороны”? Вот весьма показательный график изменения метилирования всех сотен тысяч сайтов, на которые смотрели авторы той же самой работы:

(a) Age-related changes in DNA methylation, shown as a density plot accounting for all detected CpG sites.
(b) Same as in (a), but the plots include only the CpG sites that significantly change with age.
Что мы видим? Слева вообще все сайты, а справа — только те, что значительно изменялись с возрастом. И даже если смотреть только на те сайты, что значимо изменяются с возрастом, картина не отличается кардинально. Что с возрастом глобальная картина метилирования меняется совсем незначительно: на левом графике синие кривые (молодые мыши) практически неотличимы от красных (старые мыши). Более того, основные изменения происходят в уже довольно зрелом возрасте (после 20 месяцев).
Например, на начальные участки генов (уровень метилирования которых обратно коррелирует с уровнем их экспрессии): Это ещё лучше видно, если посмотреть не просто на все сайты, а на биологически значимые из них.

Age-related changes in DNA methylation of genes. Relative position was calculated for every gene (0 corresponds to the TSS and 1 to the end of the gene) and extended in both directions up to the length of the gene. Dotted lines (individual samples) and thick lines (age groups) were calculated by generalized additive model using significant sites.
Как видно из графика выше, до этого возраста метилирование стартовых последовательностей генов практически не меняется. Возраст, когда кривые начинают сильно краснеть — это 20–22 месяцев. Да и после растёт не так сильно, как в других местах.
Вот, например, метилирование длинных некодирующих РНК (днкРНК), которые являются важным механизмом эпигенетического “тюнинга” различных клеточных процессов. А в других биологически значимых зонах видна уже более выраженная динамика. Их метилирование начинает возрастать ещё в “синем” возрасте, и изменяется куда более ощутимо:

Age-related changes in DNA methylation of certain genomic elements.
(A) Age-related changes in methylation of long non-coding RNAs. Relative positions are shown as 0 (where the element begins) and 1 (where the element ends) and extended upstream (-1) and downstream (2) according to the length of the genomic region. Lines were calculated by generalized additive model based on the significantly changed sites. Color scheme shows the age, dotted lines show individual samples, and thick lines show age groups.
У вредоносных ретротранспозонов же обратная динамика, их метилирование падает с возрастом:

Age-related changes in methylation of repetitive elements in the genome.
То есть эти древние прыгающие гены просыпаются у мышек задолго до преклонного возраста, и с каждым последующим месяцем их активность нарастает. Причём падает тоже с довольно большой скоростью и с ранним, “синим” стартом.
Но! И формально — да, часто метилирование падает или растёт в направлении среднего значения в 0,5. Потому что метилирование ДНК — процесс активный, сама по себе метильная метка на цитозин не прилепится. Если падение метилирования вполне можно объяснить пассивным процессом его “разбавления” при копировании (ну, не шмогла DNMT1 со 100%-ой точностью воспроизвести все метильные метки), то вот его рост случайным процессом объяснить сложно. Перефразируя Маяковского, если метки прилепляют, значит это кому-нибудь нужно.
Тут самое время вспомнить ту цитату авторов этой работы, которую я привёл в начале, где возрастающих сайтов метилирования было в 10 раз больше, чем падающих. Более того, мы видим, что в биологически значимых участках, например в промоторах или первых экзонах генов, рост метилирования встречается куда чаще, чем его снижение. Вот на этом весьма наглядном графике приведена динамика метилирования и других функционально значимых областей генома:

Regression slope of linear regression for every genomic region, including only the significant sites, based on the RefFreeEWAS analysis.
Хочу особо выделить те области, рост метилирования которых обычно коррелирует со снижением экспрессии их генов — промоторы, 5’UTR, и первые экзоны. Как мы видим, количество функциональных областей, где метилирование возрастает (regression slope > 0), затмевает области, где оно падает (slope < 0). Судя по другим графикам в статье, единицы измерения у этого slope — доля изменения уровня метилирования за 1 месяц жизни мыши. На графике выше их метилирование возрастает с самой большой скоростью: их regression slope равен 0,03. Отрицательное значение означает, что метилирование с возрастом снижается. То есть уровень метилирования каждого геномного участка нормализован от 0% (не метилирован совсем) до 100% (полностью заметилирован), и таким образом значение 0,03 наклона линии регрессии означает, что за 1 месяц уровень метилирования этих элементов, в среднем, возрастает на 3%, а за 30 месяцев — на 90% (то есть рост без compounding).
Быть может, это артефакт линейной регрессии для нелинейных данных — предположу, что, метилирование этих областей сильно возрастает в первые 20 месяцев жизни мышей, а в оставшиеся уже практически не меняется. Резонный вопрос — как может уровень метилирования CpG islands (островков метилирования) расти со скоростью 4% в месяц — ведь тогда за 30 месяцев средней мышиной жизни метилирование этих областей должно превысить теоретический предел в 100%.
Хотя видеть скорость роста метилирования первых экзонов в 3,5% в месяц тоже очень странно, потому что на общем графике начальных участков генов мы видим, что за всю жизнь мыши уровень их метилирования растёт не более чем на 20% — с 10% до 30% (а то и лишь до 25%, если присмотреться):

Да и на других графиках разница уровней метилирования между 3-х и 35-месячными мышами редко превышает 30%. Поэтому как в регрессии авторов статьи получились такие большие углы наклона кривых, я не очень понимаю.
А что происходит с сайтами часов метилирования?
Анализ 800 000 сайтов метилирования — это конечно, интересно, но может быть, в таком большом массиве информации мы упускаем какие-то критически важные для старения процессы? Я бы, в первую очередь, смотрел на динамику не всех сайтов, а тех, которые входят в часы метилирования, то есть сайтов, чья динамика высоко консервативна между разными животными одного возраста. Ведь в часах метилирования не 800 000 сайтов, а лишь десятки или сотни (в зависимости от часовщика), и это сайты особые, так как часто они универсальны и для быстроделящихся, и для медленно- или вообще не делящихся с возрастом клеток.
В этом, кстати, уникальность часов метилирования: самые точные из них (например, часы Хорвата) показывают практически одно и то же «время» и в нейронах, и в лимфоцитах, и в десятках других тканей:

Chronological age (y-axis) versus DNAm age (x-axis) in the test data. (A) Across all test data, the age correlation is 0.96 and the error is 3.6 years. Results for (B) CD4 T cells measured at birth (age zero) and at age 1 (cor = 0.78, error = 0.27 years), (С) CD4 T cells and CD14 monocytes (cor = 0.90, error = 3.7), (D) peripheral blood mononuclear cells (cor = 0.96, error = 1.9), (E) whole blood (cor = 0.95, error = 3.7), (F) cerebellar samples (cor = 0.92, error = 5.9), (G) occipital cortex (cor = 0.98, error = 1.5), (H) normal adjacent breast tissue (cor = 0.87, error = 13), (I) buccal epithelium (cor = 0.83, error = 0.37), (J) colon (cor = 0.85, error = 5.6), (K) fat adipose (cor = 0.65, error = 2.7), (L) heart (cor = 0.77, error = 12), (M) kidney (cor = 0.86, error = 4.6), (N) liver (cor = 0.89, error = 6.7), (O) lung (cor = 0.87, error = 5.2), (P) muscle (cor = 0.70, error = 18), (Q) saliva (cor = 0.83, error = 2.7), ® uterine cervix (cor = 0.75, error = 6.2), (S) uterine endometrium (cor = 0.55, 11), (T) various blood samples composed of 10 Epstein Barr Virus transformed B cell, three naive B cell, and three peripheral blood mononuclear cell samples (cor = 0.46, error = 4.4). Samples are colored by disease status: brown for Werner progeroid syndrome, blue for Hutchinson-Gilford progeria, and turquoise for healthy control subjects.
Ведь в нейроне и Т-клетке активны очень разные гены; более того, эти типы клеток проходят через сильно отличающееся количество делений. Для меня тот факт, что существуют универсальные эпигенетические часы для разных органов — это вообще чудо. Более того, эти эпигенетические процессы не просто синхронизированы во многих тканях нашего организма, а эта синхронность поддерживается сотню лет. И то, что и в тех, и в других с одной и той же скоростью идут некие одинаковые эпигенетические процессы — большой сюрприз! Фантастика!
Что человеческие часы метилирования весьма неплохо работают для шимпанзе (с поправкой на скорость старения, конечно). А знаете, что ещё фантастичней? Если у него получится, то такие часы вполне могут приблизить нас к ответу на любимый вопрос стохастиков: «Ну и где твои гены старения?» Потому что текущий ответ «там же, где гены эмбриогенеза и полового созревания» хоть и логически верный, с точки зрения борьбы со старением, довольно бесполезный. Стив Хорват настолько вдохновился этим, что сейчас работает над созданием универсальных часов метилирования для всех млекопитающих.
Что же происходит с сайтами часов метилирования по мере старения? Так вот. А давайте возьмём часы Хорвата и посмотрим! Соответствуют ли изменения в них тезису Александра о стохастическом дрейфе в сторону среднего? На графике ниже я построил из них кривые, отсортировав все 353 сайта часов Хорвата по медианному уровню метилирования в молодой когорте. Благо тот любезно предоставил в открытый доступ не только файл с регрессией, но и включил в него статистику по медианным уровням метилирования молодых (до 35 лет) и пожилых когорт (после 55). То есть, синяя кривая на графике ниже — это медианный уровень метилирования у молодых людей, а красная — медианный уровень метилирования тех же сайтов у пожилых:

Во-первых, видим наглядное подтверждение того, что возрастное изменение метилирования в часах Хорвата — разнонаправленное: примерно у половины сайтов метилирование возрастает, а у второй половины — убывает (осцилляция красной кривой вокруг синей). Что мы видим? Ну и в-третьих, мы видим, что у подавляющего большинства сайтов разница в уровнях метилирования между молодыми и пожилыми значениями очень мала. Во-вторых, видим, что многие возрастные изменения направлены не в сторону среднего значения метилирования (0,5), а наоборот. Чтобы лучше было видно последний тезис, вот более наглядный график абсолютной разницы медианных уровней метилирования между когортами молодых и пожилых (пересортированный по возрастанию этой самой разницы):

Как видно, для 90% сайтов абсолютный прирост или снижение метилирования не превышают 10%. Как-то не стыкуется это в моей голове со стохастическими, случайными изменениями.
Какие гены в часах меняются с возрастом больше всего?
Кстати, мне стало любопытно, а что за гены, в которых находятся оставшиеся 10% сайтов — то есть те, где метилирование сильно разнится между молодыми и пожилыми? Вот по топ-20 сайтов с каждой стороны:

Увы, мне это мало что говорит — ну да, понижается с возрастом метилирование у какого-то активатора лимфоцитов, повышается у какого-то рибосомального белка, но, в целом, всё туманно. К счастью, есть куда более умные люди, которым тоже был любопытен ответ на тот же самый вопрос, и которые провели более тщательный анализ. Это Морган Левин и её соавторы, создатели часов метилирования PhenoAge, заточенных не на хронологический возраст, а на, как они политкорректно выражаются, “фенотипический возраст”. Или, если отбросить политкорректность, на смертность. Так вот, в своих часах Морган увидела следующие функциональные изменения с возрастом:
Эти пути включали (но не ограничивались ими): множественные сигнальные пути toll-подобных рецепторов (7, 9, 3, 2), регуляцию воспалительного ответа, каскад JAK-STAT, ответ на липополисахарид, сигнальный путь TNF, и положительную регуляцию активности транскрипционного фактора NF-kappaB. Среди тех каскадов, у которых была положительная корреляция со старением (повышенная экспрессия у более эпигенетически старых людей), мы наблюдали ряд провоспалительных сигнальных путей. Другие интересные положительно ассоциированные гены включали: ответ на питательные вещества, каскад JAK-STAT, участвующий в сигнальном пути гормона роста, многоклеточный рост организма и регуляцию метилирования ДНК. Кроме того, среди положительно ассоциированных генов были каскады антивирусного ответа — каскад интерферона типа I, защитный ответ на вирусы, интерферон-гамма-опосредованный сигнальный путь, клеточный ответ на интерферон-альфа и т.д.
К ним относятся: инициация трансляции; регулирование инициации трансляции; сборка большой рибосомальной субъединицы; сборка малых рибосомальных субъединиц; трансляционная элонгация; инициация транскрипции с промотора РНК-полимеразы I; связанная с транскрипцией нуклеотид-эксцизионная репарация; нуклеотид-эксцизионная репарация; распознавание повреждений ДНК; реакция на повреждение ДНК, обнаружение повреждений ДНК; и регулирование чекпоинта повреждений ДНК. При тестировании на обогащение среди генов, которые отрицательно коррелировали с DNAm PhenoAgeAccel (сниженная экспрессия у более эпигенетически старых людей), мы обнаружили, что многие из них были вовлечены в процессы, включающие транскрипционные и трансляционные механизмы, а также распознавание и восстановление повреждений.
Здесь мой взгляд зацепился за следующее: то, что с возрастом растёт хроническое воспаление известно давно, а вот увидеть ещё одно подтверждение глобальной активации интерферонового ответа 1-го типа было весьма интересно. Ведь этот ответ заточен на борьбу с вирусами («ахтунг, ДНК в цитоплазме!»), и есть свидетельства (1, 2, 3) того, что тут речь идёт не столько о внешних вирусах, сколько о внутренних — ретровирусах-транспозонах, обитающих в нашем геноме, и активирующихся где-то после 45–50 лет. Андрей Гудков даже придумал термин для этой братии — «ретробиом», и вместе с коллегами показал, что если подавить активность ретроэлементов с помощью ингибиторов обратной транскриптазы (которая и создаёт в цитоплазме ДНК этих ретроэлементов), то интерфероновая активность значительно снижается. Вообще транспозоны — это отдельная песня, заслуживающая свою собственную статью.
Часы часам рознь
Кстати, стоит оговориться, что различных человеческих часов метилирования уже существует более десятка, и они довольно разнородны. Уже упомянутая выше Морган Левин с соавторами провела отличный анализ эпичасов. Как видно из её таблицы ниже, какие-то из часов изначально были заточены на корреляцию с паспортным возрастом, какие-то на смертность. В каких-то сотни сайтов метилирования, в других — единицы:

Одни показывают высокий уровень синхронизации между тканями (исходные часы Хорвата тут пока лидируют), другие — низкий:

Как мне кажется, это отражает различные возрастные эпигенетические процессы: наверняка, есть какие-то тканеспецифичные эпигенетические процессы старения, а есть и глобальные. Разные часы выявляют разные аспекты этих процессов. И, конечно же, мы пока на ранних этапах пути к разгадке биологических смыслов этих процессов. Но я уверен, что эта разгадка не за горами.
Отключение нужных генов с возрастом
Возвращаясь к функциональным возрастным изменениям, во второй части приведённой выше цитаты от Морган Левин мне было интересно в очередной раз увидеть свидетельство того, что с возрастом у людей эпигенетически снижается активность каскадов распознавания повреждений ДНК и их починки. Кстати, это контрастирует с данными на мышах, где группа Гладышева видела обратную динамику.
По этой теме возрастного снижения рибосомальной функции мне в память врезался постер московской группы всё того же Вадима Гладышева с конференции в Казани в 2018 году. А вот в чём мыши и люди сходятся, так это в том, что с возрастом эпигенетически снижается активность механизмов транскрипции и трансляции ДНК, включая построение рибосом. Вот цитата из их абстракта с той конференции:
Неожиданно было видеть, что [в результатах рибосомального профайлинга клеток после фиксации] с возрастом падало количество рибосом на старт-кодонах, а росло на стоп-кодонах, что соответствовало системной деградации протеостаза по мере старения. Мы наблюдали возрастное снижение уровней экспрессии многих генов, ассоциированных с трансляцией, включая гены, вовлечённые в рибосомальный биогенез и рекрутирование мРНК.
Можно подумать, возрастное снижение мышиной рибосомальной функции изучает только одна группа Гладышева, но нет, не Гладышевым единым: есть работы и других групп, показывающие, что с возрастом снижается активность построения рибосом и генной трансляции, в целом. Вот, например, работа на мышах, показавшая возрастное снижение экспрессии не только рибосомальных, но и митохондриальных генов (выделены красным ниже) в мышиных кардиомиоцитах:

А совсем недавно вышла очень красивая работа на крысах, где авторы проследили возрастные изменения генной экспрессии в 4 типах тканей: печень, почки, мышцы (икроножные) и мозг (гиппокамп). В этой работе тоже прослеживаются схожие темы: активация генов врождённого иммунитета (блок № 1 на графике ниже) и снижение генов митохондриальной функции (блок № 7):

При этом интересно наблюдать, что генная экспрессия в гиппокампе не так уж сильно изменялась по сравнению с остальными тремя типами ткани. Похоже, мозг или, по крайней мере, гиппокамп, стареет медленнее, чем другие органы. А вот в мышцах и почках гены апоптоза значительно увеличивают свою активность в пожилом возрасте, что может объяснить и возрастную саркопению, и почечную недостаточность. Вот как авторы сами резюмируют свои находки:
Возрастная регуляция этих путей также наблюдалась в гиппокампе, хотя и менее выраженно. Наиболее поразительно, что пути, которые были связаны с активацией воспаления, были доминирующей темой, которую мы наблюдали; например, пути, связанные с врожденным иммунным ответом, воспалением и передачей сигналов цитокинами, с возрастом повышались в печени, икроножных мышцах и почках. Система комплемента также повышала активность с возрастом во всех четырех тканях. Пути, связанные с отторжением аллотрансплантата и альфа- и гамма- интерфероновыми ответами, сильно возрастали с возрастом в почках, печени и икроножной мышце, а также в гиппокампе. Заметное подавление окислительного фосфорилирования и дыхательного электронного транспорта наблюдалось в икроножной мышце. В дополнение к воспалению, меняли активность и другие пути, представляющие интерес; апоптозный каскад повышался в печени, икроножной мышце и почка, и в меньшей степени в гиппокампе, что указывает на общее увеличение клеточной гибели в тканях с возрастом.
…
Наиболее заметно подавляемыми по мере старения оказались следующие каскады: митохондриальный; окислительное фосфорилирование, транспорт дыхательных электронов и биологическое окисление — все они постепенно снижались с возрастом в печени и почках. Эти изменения согласуются с идеей, что митохондрии становятся менее компетентными с возрастом, лишая клетки критических запасов АТФ, а также множества митохондриальных сигналов. Возрастная регуляция окислительного фосфорилирования, дыхательного электронного транспорта и путей биологического окисления не была заметна в гиппокампе.
Есть и другие свидетельства того, что с возрастом отключаются нужные гены. В своё время мне очень понравилась эта статья, в которой авторы проанализировали метилом и транскриптом лейкоцитов, и смогли найти в этих клетках те гены, чья экспрессия коррелировала с изменениями их метилирования. Как уже было показано в других работах, многие из значительно меняющихся с возрастом генных каскадов связаны с иммунной системой:
Мы выявили 20 GO областей, из которых 6 (30%) были связаны с иммунной системой. Чтобы проанализировать процессы, связанные с генами, которые показали корреляцию между уровнями экспрессии и метилирования, мы выполнили анализ генной онтологии для 80-летних. Только один функциональный термин GO (GO: 0005515 Белковое связывание) был связан с коррелированными CpGs [сайтами метилирования]. Многочисленные пути иммунной системы были также выявлены при рассмотрении GO процессов, которые были более слабо связаны с этими генами, где 39 из 121 (32%) статистически значимых терминов GO-процесса были связаны с иммунной системой. Анализ канонических путей выявил 15 канонических путей, большинство из которых были напрямую связаны с иммунной системой (коммуникация между дендритными клетками и клетками-киллерами, путь антиген презентации, рецептор Fcγ, опосредованный фагоцитоз в макрофагах и моноцитах, дифференцировка Т-хелперов) или связаны с ремоделированием цитоскелета и эндоцитозом (интегрин-сигнализация, актиновая сигнализация цитоскелета, Tec-киназная сигнализация, сигнализация паксиллина, кавеолярно-опосредованная сигнализация эндоцитоза). В дополнение к иммунной системе были затронуты пути, связанные с реакцией на окружающую среду.
Но больше всего мне понравился выделенный жирным вывод авторов, потому что он полностью совпадает с моим пониманием:
Наши результаты также показывают, что гипо- и гиперметилирование, связанные со старением, являются различными процессами: гиперметилирование может быть вызвано запрограммированными изменениями, тогда как гипометилирование может быть результатом экологических и стохастических процессов. Мы считаем, что определенные возрастозависимые нарушения иммунной системы могут быть опосредованы изменениями в метилировании ДНК.
Что-то похожее увидели и авторы этой очень подробной работы:
BACH2 играет решающую роль в опосредованных Т-клетками иммунных реакциях.
…
Этот факт наводит на мысль, что даунрегуляция BACH2 может быть связана с сенесценцией Т-клеток, и может контролироваться гиперметилированием ДНК в локусе BACH2 в дополнение к модификациям гистонов. Примечательно, что мы наблюдали повышенный уровень метилирования ДНК в локусе BACH2 и пониженную экспрессию BACH2 в CD4 + T-клетках в группах среднего возраста и долгожителях по сравнению с группой новорожденных.
Кстати, раз уж зашла речь о роли эпигенетики в кроветворении, не могу не упомянуть другую мою любимую работу. В ней авторы выключили в гемопоэтических стволовых клетках мышей ген DNMT3A, то есть ген одной из метилаз — ферментов, выполняющих метилирование ДНК. И эти клеточные линии стали, по сути, бессмертными:
Молекулярная характеристика показывает, что эта иммортализация in vivo связана с постепенными и очаговыми потерями метилирования ДНК в ключевых регуляторных областях, связанных с генами самообновления. Здесь мы показываем, что Dnmt3a-нокаутные гемопоэтические стволовые клетки могут регенерировать по меньшей мере в течение 12 поколений трансплантаций у мышей, тем самым значительно превышая продолжительность жизни нормальных ГСЭ.
То есть после подавления активности одной из метилаз, в иммортализированых таким образом клетках авторы увидели снижение метилирования в регуляторных участках генов, отвечающих за обновление этих самых клеток. Закрадывается резонный вопрос: а почему в обычных мышах эти важные гены метилируются с возрастом? Эти наблюдения наводят на мысль, что та деградация стволовых гемопоэтических клеток, которую мы наблюдаем по мере старения, вполне может быть вызвана не внешними факторами (износ, усталость от жизни), а внутренними — возрастной эпигенетической программой.
Там тоже есть интересные наблюдения. А что в других тканях? Вот, например, какие эпигенетические изменения увидела группа немецких исследователей в тканях кишечника у мышей (клетках кишечных крипт и стволовых клеток кишечника):
Эти данные указывают на то, что зависимое от старения гиперметилирование CpG-островков может играть ключевую роль как в возрастной потере функциональности кишечника (путем подавления факторов, необходимых для поддержания структуры кишечника и способности к дифференцировке), так и в развитии рака. Мы обнаружили гиперметилирование как генов, важных для гомеостаза кишечника (например, Wnt3a, Cbx6, Pak3, Nr5a2), так и генов, часто эпигенетически подавленных при раке (например, Cdk, Dkk, семейство генов Sfrp).
В общем, мне кажется, уже накоплен довольно большой массив данных, указывающий на важнейшую роль эпигенетики как драйвера многих возрастных патологий.
Не метилированием единым
Метилирование ДНК — регуляторная фича генных сетей млекопитающих и прочих позвоночных. Но есть виды, у которых метилирования ДНК либо вообще нет, либо регуляторной роли в эпигенетике оно не играет. Зато у этих видов есть гистонная регуляция, которая, конечно же, есть и у нас, и обычно синхронизирована с нашим метилированием (репрессированные конфигурации метилирования совпадают с репрессированными конфигурациями гистонов, и наоборот, активная конфигурация метилирования обычно совпадает с активными гистонами):

Targeting of DNA methylation and H3K9 methylation to (a) heterochromatin, where it interacts with the H3K9 methylation system, and (b) gene bodies, where it may interact with H3K36 methylation to target DNA methylation to these regions. Several alternative but not mutually exclusive models are shown here.
Кстати, тот факт, что гистонная регуляция есть у всех эукариотов (включая одноклеточных дрожжей), а метилирование далеко не у всех, наводит меня на мысль, что метилирование — гораздо более молодой эволюционный механизм эпигенетического контроля.
Что я хотел показать, так это то, что даже у гораздо более «примитивных» видов, чем мы или мыши, прослеживается эпигенетическое отключение нужных генов с возрастом. Но я отвлёкся. В ней авторы показали, что у нематод эпигенетически отключается каскад теплового шока — один из главных механизмов контроля качества белков. Один из моих любимых примеров — статья про нематод с игривым названием «Шокирующе рано: хроматин-опосредованная потеря каскада теплового шока». Причём отключается внезапно и в довольно раннем возрасте: всего в течение 4-х часов после полового созревания:
У метазоа старение связано со снижением контроля качества, что повышает риск возникновения конформационных заболеваний белка. Реакция на тепловой шок (HSR) имеет важное значение для протеостаза и клеточного здоровья. elegans HSR резко снижается в течение 4-часового периода в раннем взрослом возрасте, совпадающего с наступлением репродуктивной зрелости. Здесь мы показываем, что у C.
1. Репрессия HSR происходит из-за увеличения меток H3K27me3 в локусах генов стресса, тайминг которых совпадает со сниженной экспрессией деметилазы H3K27 jmjd-3. 2F и S2E). Это приводит к репрессированному состоянию хроматина, которое препятствует связыванию HSF-1 и подавляет инициацию транскрипции в ответ на стресс.
…
Мы обнаружили, что индуцируемость HSR снижается на 60–70% между 8 и 12 часами в первый день взрослой жизни и совпадает с началом яйцекладки (рис. Кроме того, быстрая репрессия HSR коррелирует с резким ухудшением способности животных восстанавливаться после острого теплового шока (рис. Это говорит о том, что репрессия HSR представляет собой активно контролируемую транскрипционную перенастройку, которая отмечает начало репродуктивной зрелости и может являться первым молекулярным событием в процессе старения. 2G), подтверждая, что переключение транскрипции в середине первого дня взрослой жизни имеет глубокие последствия для организма.
…
Наши данные свидетельствуют о том, что пути реакции на стресс не дерегулируются постепенно как следствие стохастических повреждений с возрастом, а вместо этого быстро и точечно подавляются, когда животные начинают размножаться.
Ну чем не феноптоз? Кстати, весьма схожее эпигенетическое отключение систем протеостаза (поддержания качества белков), только уже у коловраток, наблюдали и авторы этой работы:
manjavacas. Снижение белкового гомеостаза (протеостаза) считается одним из признаков старения среди многих таксонов, и наши результаты указывают на такое снижение в поздней жизни у B. 8); Результаты GSEA фиксируют снижение экспрессии генов протеасом в позднем возрасте (Рис. Экспрессия почти всех генов, связанных с протеасомами, плавно увеличивается в течение жизни вплоть до перехода к репродуктивному старению, после чего экспрессия 31 из 38 аннотированных структурных субъединиц генов протеасомы и её каталитической субъединицы снижается в 2,4 раза (рис. 5).
…
Вместе эти результаты позволяют предположить, что большие изменения в эпигенетических маркерах могут быть частично или даже в значительной степени ответственны за управление изменениями экспрессии генов в течение продолжительности жизни, и эта идея все чаще подтверждается результатами других модельных систем.
Кстати, этот упомянутый в цитате Рис. 5 вообще очень наглядно показывает, сколько ключевых систем снижает свою активность под конец репродуктивного периода:

При этом после окончания репродукции коловратки живут ещё 20-30% своей продолжительности жизни, а нематоды вообще все 80% — фертильный период у нематод длится лишь 3-4 дня, после чего черви остаются в живых ещё 2-3 недели. Почему гены начинают отключать у них не только важные системы гомеостаза, но и саму репродуктивную систему так рано? Ведь теоретически, с точки зрения индивидуального отбора, продление способности плодиться давало бы им эволюционное преимущество. Видимо, всё же есть давление отбора на более высоких уровнях фрактальности, чем уровень особи, которое и закрепляет как феноптоз, так и раннюю стерилизацию в таких видах.
Альцгеймер тоже обусловлен эпигенетикой?
Продолжая тему эпигенетически обусловленной возрастной деградации, не могу не упомянуть недавнюю работу, которая даже для такого заядлого «программиста» как я стала неожиданностью. В ней американские исследователи показали, что болезнь Альцгеймера может быть эпигенетически обращена вспять. Если честно, я думал, что Альцгеймер обусловлен необратимой гибелью нейронов, и что его можно только профилактировать, но не лечить. К счастью, это может оказаться не так.
Глутамат — это основной нейромедиатор, а его рецепторы играют важную роль в процессах памяти. Авторы вышеупомянутой работы заметили, что у больных Альцгеймером эпигенетически подавляется активность глутаматных рецепторов. Следующим логическим шагом было попробовать заблокировать этот репрессивный механизм и посмотреть, что получится. Так вот, исследователи установили, что это подавление происходит через один из гистонных механизмов генной репрессии.
Показатели когнитивных тестов взмывали ввысь после введения ингибитора генной репрессии: А получилось чудо: у мышей со значительными дефицитами памяти после введения ингибиторов того самого репрессивного эпигенетического механизма память восстанавливалась практически до уровня здоровых животных.

FAD mice exhibited deficits in novel object recognition memory, working memory and spatial memory, which was rescued by EHMT1/2 inhibitors.
(A) Bar graphs (mean ± SEM) showing the discrimination ratio of novel object recognition (NOR) tests in wild-type (WT) versus FAD mice (5–6 months old) without or with the treatment of BIX01294 (1 mg/kg, s.c. 3x) or UNC0642 (1 mg/kg, i.p. 3x). **P < 0.01, one-way ANOVA. (B) Representative heat maps illustrating the time spent in different locations of the arena for novel object recognition tests of all groups (blue: 0 s; red: ~10 s). Locations of novel (denoted by an arrow) and familiar objects are labelled with the circles or squares. (С) Scatter plots showing the discrimination ratio of novel object recognition tests in each of the examined wild-type or FAD mice before and after the treatment with BIX01294 or UNC0642. *P < 0.05, ***P < 0.001, paired t-test. (D) Bar graphs (mean ± SEM) showing the percentage correctness in T-maze working memory (WM) tests in wild-type or FAD mice with or without BIX01294 treatment. *P < 0.05, **P < 0.01, two-way ANOVA. (E) Scatter plots showing the percentage correctness in T-maze tests in each of the examined FAD mice before and after BIX01294 treatment. ***P < 0.001, paired t-test. (F) Representative heat maps illustrating the time spent in different locations of the arena for Barnes maze tests during the memory phase (escape box removed) in wild-type versus FAD mice without or with the treatment of BIX01294 (1 mg/kg, s.c. 3x) or UNC0642 (1 mg/kg, i.p. 3x) (blue: 0 s; red: ~10 s). Locations of the correct (indicated by an arrow) and seven incorrect holes are labelled with circles. (G) Bar graphs (mean +/ SEM) showing the time spent on exploring the correct hole (T1) versus the seven incorrect holes (T2) in the memory phase of Barnes maze tests of all groups. *P < 0.05, **P < 0.01, ***P < 0.001, two-way ANOVA. (H) Bar graphs (mean +/ SEM) showing the spatial memory index (T1/T2) of Barnes maze tests in wild-type versus FAD mice without or with the treatment of BIX01294 or UNC0642. ***P < 0.001, two-way ANOVA. (I) Scatter plots showing the spatial memory index in Barnes maze tests in each of the examined FAD mice before and after the treatment with BIX01294 or UNC0642. **P < 0.01, ***P < 0.001, paired t-test. (J) Plots (mean +/ SEM) of spatial memory index in FAD mice treated with BIX01294 (1 mg/kg, s.c. 3x) or saline at different time points. **P < 0.01, ***P < 0.001, saline versus BIX01294; ##P < 0.01, ###P < 0.001, pre- versus post-injection, two-way ANOVA. Each set of the experiments was replicated between four and five times.
Но самое главное — это то, что мы увидели первые звоночки совершенно потрясающей перспективы: Альцгеймер может быть эпигенетически обратим. Правда, действие этой терапии было довольно краткосрочным: спустя 7 дней после введения препаратов память мышей опять ухудшалась до исходных показателей, что наводит на мысль, что именно столько времени уходит у того гистонного репрессивного механизма на восстановление «плохого» эпигенетического профиля активности генов глутаматных рецепторов.
Эпигенетический откат
Ну что ж, думаю, я привёл достаточно примеров того, как много плохих вещей, происходящих с организмом по мере старения, обусловлены эпигенетическими механизмами. Напоследок лишь напомню, что в этом есть и хорошая новость — эпигенетика обратима! Самый яркий пример этого — эпигенетический откат с помощью репрограммирования и сопутствующее ему функциональное омоложение клеток. Факторы Яманаки способны омолодить даже клетки от столетних доноров: например, восстановить у них митохондриальную функцию.
Например, в этом препринте это было продемонстрировано на человеческих фибробластах — а именно, что снижение их эпигенетического возраста (синяя кривая на графике ниже) идёт плавно, так же как и снижение экспрессии функциональных генов, грубо говоря, отвечающих за то, что фибробласт остаётся фибробластом: Более того, нам крупно повезло, что факторы Яманаки снижают эпигенетический возраст постепенно.

Открытие этого постепенного свойства эпигенетического репрограммирования позволяет надеяться на то, что нам удастся найти безопасное терапевтическое окно (например, отмеченное желтой рамкой на графике выше) — то есть период безопасного эпигенетического отката, когда клетка уже эпигенетически омолодилась, но ещё не потеряла своих функциональных характеристик (т.е. клетка кожи остается клеткой кожи, а не дедифференцируется в плюрипотентную клетку).
Его приводят в своих презентациях и такие мэтры как Стив Хорват или Майк Уэст. Кстати, в вышеприведённый график плавного отката не я один влюбился с первого взгляда. Это и выходцы из Стэнфорда Turn Biotechnologies, и перепрофилировавшийся в эпиоткат AgeX со своим REVERSE Bioengineering, и гарвардец Синклер, который ещё вчера продвигал ресвератрол и никотинамид, а сегодня уже выпускает книгу, где говорит, что старение обусловлено эпигенетикой, и активно готовится проводить клинические исследования по эпиоткату. И да, приятно видеть, что с тех пор как я загорелся темой эпиотката в далёком 2017 году, полку эпиоткатчиков прибыло.
А вдруг эпичасы отражают лишь поломки ДНК?
Правда, несмотря на то, что я разделяю оптимизм Синклера по поводу ценности частичного репрограммирования для эпигенетического омоложения, я категорически не согласен с его видением механизмов этого процесса. В своих выступлениях и недавнем препринте (в соавторстве с Вадимом Гладышевым, кстати), он говорит о старении как о потере эпигенетической информации из-за накопления эпигенетического шума, а о репрограммировании как о восстановлении «правильных» эпигенетических настроек из какого-то информационного бэкапа.
Мол, сайты метилирования, по которым строятся эпигенетические часы, — ничто иное как просто сайты починки двуцепочных разрывов, которые рандомно появляются с возрастом. В рамках этой парадигмы Синклер и его сторонники полагают, что эпигенетические часы лишь отражают стохастические поломки нашей ДНК.
Как минимум, по двум причинам. Сразу скажу, что мне этот тезис видится ложным. У половины сайтов оно растёт, у другой половины — падает. Во-первых, метилирование сайтов в эпичасах изменяется разнонаправленно. Во-вторых, как-то наивно предполагать, что с возрастом ДНК ломается у всех нас в одних и тех же сайтах одновременно. Какой из этих процессов должен отражать починку ДНК в вышеозвученном тезисе? Ведь как я уже упоминал, сайты в часах метилирования Хорвата универсальны для 51 типа клеток: как быстроделящихся, так и не делящихся после рождения вовсе. Да ещё и в разных тканях: что в мозге, что в крови.
В этой работе они создали трансгенных мышей, у которых можно было в любой момент вызывать разрывы ДНК, и попытались показать, что избыточные разрывы ДНК увеличивают эпигенетический возраст. Но в поддержку своего тезиса Синклер и соавторы недавно выпустили ещё один препринт с говорящим названием «Эпигенетический дрейф, вызванный разрывами ДНК, как причина старения млекопитающих».
Причем при активации она разрезала ДНК не абы где, а в строго определённых 20-и сайтах (красные точки на картинке ниже): Чтобы вызывать такие разрывы не случайным образом, а on demand, авторы внедрили в геном мышей специальную генную кассету с «генными ножницами», которая по дефолту была не активна, а запускалась, если мышам давали молекулу-активатор.

Никаких данных по влиянию таких разрывов ДНК на срок жизни своих мышей авторы не приводили. Так вот, свой ключевой тезис, что именно двуцепочные разрывы вызывают старение, авторы базировали лишь на том, что часы метилирования (да и то, лишь в культуре клеток) у таких мышей показывали более позднее время. Даже наоборот, они цитировали другую работу, которая показала, что мыши с повышенным мутагенезом живут столько же, сколько и обычные.
Хоть они и говорят, что после починки разрывов ДНК у мышей в 1,5 раза был выше эпигенетический возраст, при ближайшем рассмотрении оказывается, что в 1,5 раза — это на каких-то никому не известных часах (левый график ниже), а вот на часах Гладышева (правый график), разница в часах метилирования была лишь в районе 10%. Кстати, и с самими часами метилирования у авторов не всё было гладко. При этом эпивозраст группы контроля различался между двумя этими часами в 2,5 (!) раза:
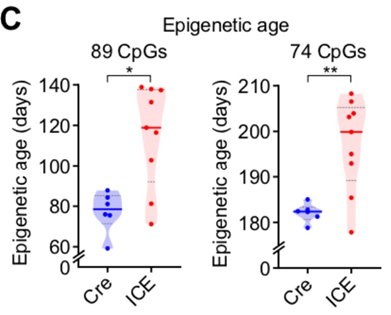
Ну и напоследок, видимо, чтобы окончательно потопить свой же тезис, авторы сами признают, что сайты их индуцируемых двуцепочных разрывов совсем не совпадают с сайтами метилирования их эпичасов. Это наглядно видно на предыдущем графике с хромосомами и отмеченными на них сайтами метилирования (синим) и сайтами разрывов (красным).
Мол, при разрыве ДНК репрессивные комплексы безвозвратно перемещаются с сайтов метилирования на места разрывов для их починки. В свете этих фактов выдвинутый авторами предполагаемый механизм влияния разрывов ДНК на метилирование выглядит совсем слабо. Более того, объяснить снижение метилирования с возрастом можно и проще: просто стохастическим размыванием при делении клеток. Но это плохо сочетается с тем, что, как я уже сказал, метилирование сайтов эпичасов меняется разнонаправленно. Ведь новое метилирование — энергозатратный процесс, который вряд ли будет происходить случайно в одних и тех же сайтах у всех особей одного возраста. Но вот как объяснить его рост?
Я считаю, что факторы Яманаки возвращают назад эпигенетические настройки не потому, что молодые настройки тайным образом записаны где-то в бэкапе, а потому что откат эпигенетической программы старения как начальный этап дедифференциации — явно заложенный в наши гены механизм. Поэтому мне видится гораздо более правдоподобной совсем другая природа часов метилирования и процесса эпиотката. Вообще, у бессмертной половой линии есть ещё пара трюков по своему омоложению, благодаря которым старая яйцеклетка уже миллионы лет без проблем превращается в новую особь — надеюсь, что когда-то я так же превращу свою двухчасовую лекцию по этой теме в полноценную статью. Быть может, тот же самый механизм используется для омоложения яйцеклетки после оплодотворения.
Возвращаясь к моему любимому эпиоткату, сам факт того, что он возможен, и происходит плавно, мне говорит о том, что снижение экспрессии полезных для нашего организма генов с возрастом тоже эпигенетически запрограммировано, а не происходит случайным образом из-за какого-то «зашумления». Но я опять отвлёкся. Прямо как в анекдоте про покойника, 40 раз подряд поскользнувшегося на нож. Ну что это за зашумление такое, происходящее у всех одинаковым образом?
Как и не было бы эпигенетического отката: ведь репрограммирование — явно плавный процесс, заканчивающийся полной потерей дифференциации и превращением клетки в плюрипотентную. Ведь если бы это был случайный процесс, никаких эпигенетических часов у нас бы не было, так как слишком большой был бы разброс между людьми, причем с возрастом только нарастающий. То есть старый фибробласт намного ближе к молодому фибробласту по своей эпигенетике, чем к плюрипотентной клетке. Более того, эпигенетические настройки конечной плюрипотентной (считай, эмбриональной) клетки явно отличаются от настроек «молодой» дифференцированной клетки того типа, которым она была перед тем как была перепрограммирована. И какие из них считать «молодыми»? Так какие именно из этих «молодых» эпинастроек записаны в бэкапе? Сразу после? До полового созревания?
Старение — программа. Короче. Но это не страшно, ведь нет ничего постыдного в заблуждениях. И только нежелание признать очевидное вынуждает стохастиков придумывать невероятные гипотезы, чтобы утрамбовать эпигенетический откат этой программы в свою картину мира. К полной победе над старением. Главное — учиться на ошибках, и шаг за шагом продолжать двигаться вперёд. Аминь. Программой.

![Фото [Перевод] Как я выиграл Хакатон, едва не потеряв рассудок](http://orion-int.ru/wp-content/uploads/2024/03/xperevod-kak-ya-vyigral-xakaton-edva-ne-poteryav-rassudok-390x220.png.pagespeed.ic.uqlKF6sbRM.jpg)




